Understanding
the genetic architecture of
human complex traits
Most human traits, including common diseases (e.g., obesity, cardiovascular disease, cancer, and mental illness), are complex because they are often affected by many genetic and environmental factors. Understanding the genetic architecture of complex traits and mapping the relevant genes are fundamental goals in genetics and medical research.
As a gene-mapping technology, genome-wide association study (GWAS) has revolutionized the field of human genetics by the identification of tens of thousands of genetic variants associated with thousands of human traits. Nevertheless, GWAS was initially criticized for only explaining a small fraction of the heritability known from family-based studies, i.e., the so-called “missing heritability” puzzle (Manolio et al. 2009 Nature). We developed a method (called GREML) to repurpose GWAS data to estimate heritability in unrelated individuals (Yang et al. 2010 Nat Genet). We showed that a large proportion of the heritability for human height could be explained when considering the single nucleotide polymorphisms (SNPs) available on a commercial SNP array altogether, implying that heritability is not missing and that most of the trait-associated SNPs remained undetectable primarily because the experimental sample sizes were not large enough. The GREML method has subsequently been extended to partition the SNP-based heritability into components attributed to individual chromosomes (Yang et al. 2011 Nat Genet) or sets of genetic variants stratified by minor allele frequency (MAF) or linkage disequilibrium (LD) properties of the variants (GREML-LDMS; Yang et al. 2015 Nat Genet).
Further extension of the GREML model in a Bayesian framework leads to the development of the BayesS method that allows for the estimation of three important genetic architecture parameters simultaneously, i.e., the SNP-based heritability, the polygenicity (proportion of variants with non-zero effects), and the relationship between MAF and effect size (Zeng et al. 2018 Nat Genet). Along with evidence from other studies (O'Connor et al. 2019 AJHG), our research suggests that polygenic architecture is the norm for most human traits, likely to be a consequence of purifying selection (a.k.a. negative selection; Zeng et al. 2018 Nat Genet; Zeng et al. 2021 Nat Comm). Future research directions include estimating SNP-based heritability using whole-genome sequence data (Wainschtein 2021 Nat Genet), disentangling the genetic architecture of molecular phenotypes in normal and tumor tissues (e.g., Sun et al. 2021 Cancer Res), and quantifying phenotypic variance explained by ultra-rare and structural variants.
Methods for
genome-wide association
(GWA) analysis
Population stratification and relatedness are two major confounding factors that could cause inflation of test statistics in GWAS. Linear mixed model (LMM)-based association methods that fit many variants as random effects to control for background genetic effects when testing a variant in query for the association have been proved effective in capturing known and unknown confounding effects. We pointed out that the conventional LMM-based association methods are underpowered because of double fitting (i.e., fitting the variant in query twice, once as a fixed effect and again as a random) and proposed the leave-one-chromosome-out (LOCO) strategy as a remedy (Yang et al. 2014 Nat Genet).

On the other hand, computing time is always the biggest challenge for the LMM-based association methods owing to the computational complexity of handling the random effects. We tackled this issue by developing fastGWA, an ultra-efficient LMM-based association test that can analyze the whole UK Biobank data (n ~= 400,000) in less than 30 minutes (Jiang et al. 2019 Nat Genet). The fastGWA method has recently been extended to deal with highly unbalanced case-control data, i.e., the fastGWA-GLMM method (Jiang et al. 2021 Nat Genet). Both fastGWA and fastGWA-GLMM are scalable to GWAS data with sample sizes of over a million.
Another direction of interest is the development of methods for the re-analysis of GWAS summary statistics. One example is the COJO method that utilizes summary statistics from a GWAS and LD information from a reference sample for a conditional or joint multi-variant association analysis without the need to access the individual-level data of the discovery GWAS sample (Yang et al. 2012 Nat Genet). This opens a new avenue for developing summary data-based methods for the estimation and partition of heritability, fine-mapping, gene-based tests, and polygenic risk prediction.
From GWAS to biology
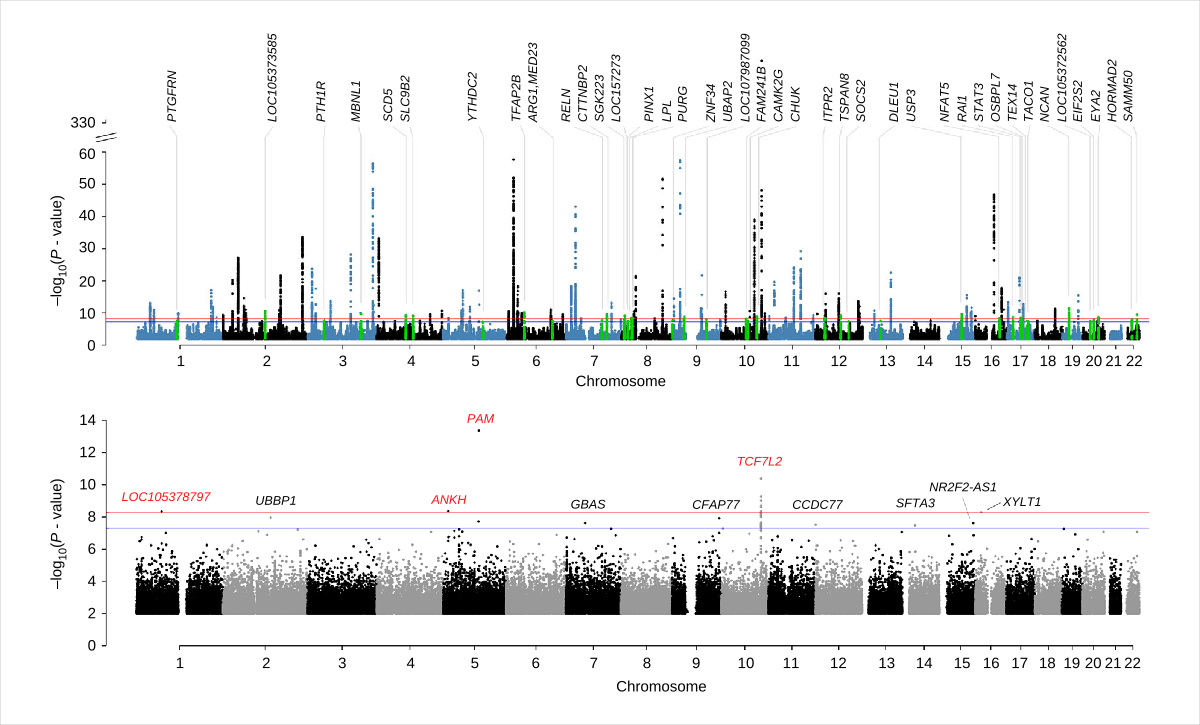
The association signals identified from GWAS are informative in terms of the genomic locations of the loci conferring the traits. However, due to the complexity of LD between genetic variants, the causative variants underpinning the GWAS signals are often elusive, and the genes on which the causal variants act are also largely unknown. We have a long-standing interest in developing analytical methods, curating relevant data, and performing integrative analyses to identify genes and/or functional genomic elements responsible for the GWAS signals for a range of complex traits and disorders such as obesity and type-2 diabetes.
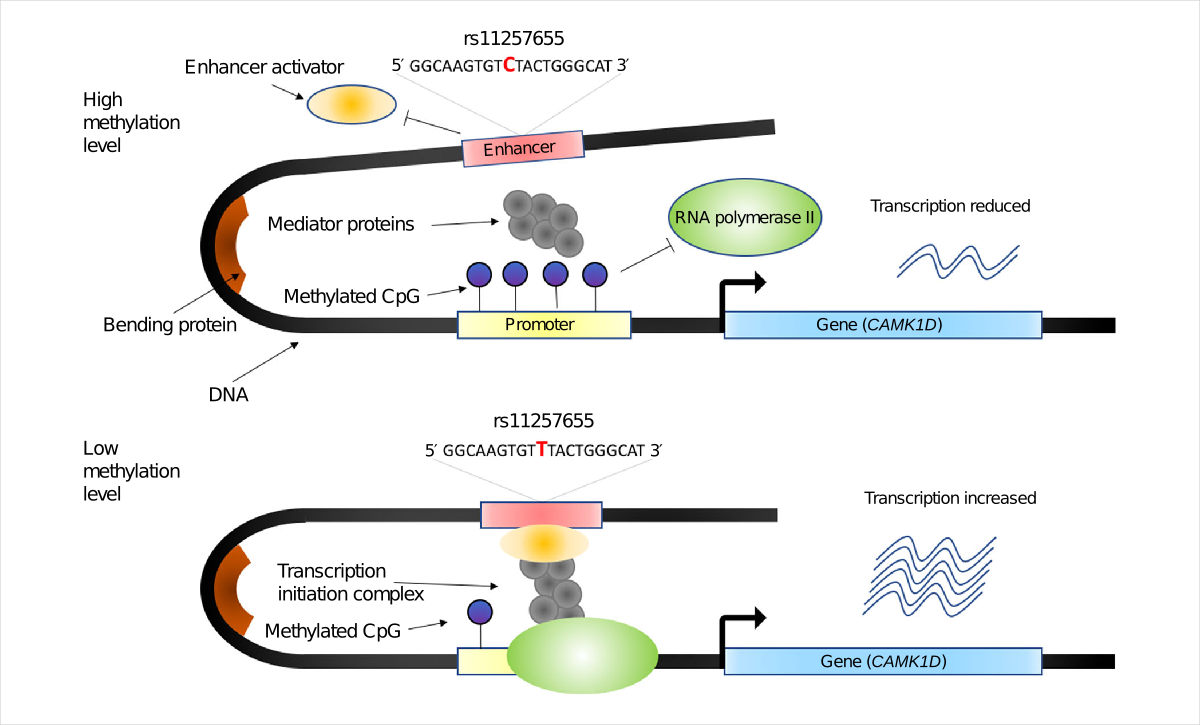
We have developed a set of methods, SMR (summary-data-based Mendelian randomization) and HEIDI (heterogeneity in dependent instruments), to integrate GWAS and expression quantitative trait locus (eQTL) data for identifying genes whose transcriptional levels are associated with a trait of interest because of a set of shared causal variants (Zhu et al. 2016 Nat Genet). The SMR and HEIDI methods have subsequently been applied to identify associations between DNA methylation (DNAm) sites and a trait of interest and between DNAm and gene expression (Wu et al. 2018 Nat Comm). Further incorporation of chromatin accessibility and histone modification data from epigenomic studies facilitated the inference of molecular mechanisms whereby the genetic variants exert their effects on disease risk (Wu et al. 2018 Nat Comm; Xue et al. 2018 Nat Comm).
Moving forward, we are interested in understanding the spatio-temporal effects of trait-associated variants (e.g., the genetic effect correaltion between brain and blood; Qi et al. 2018 Nat Comm), genotype-by-environment interaction (e.g., via the variance quantitative trait locus, vQTL, analysis; Yang et al. 2012 Nature; Wang et al. 2019 Science Advances), understanding the role of other gene regulation mechanisms in mediating the GWAS effects (e.g., mRNA splicing; Qi et al. 2021 Nat Genet), and developing methods that can integrate multiple types of omics data jointly to explain the GWAS signals. A key focus is to pinpoint the specific cellular contexts of genetic effects, which has led to the development of methods like MeDuSA, for deconvolving cell-state abundances from bulk tissue data (Song et al. Nat Comput Sci 2023), and gsMap, a novel method that integrates GWAS summary statistics with high-resolution spatial transcriptomics data to map trait-associated cells in their native tissue context (Song et al. 2025 Nature).
Developing
bioinformatics tools
We reason that any useful statistical method should be implemented in easy-to-use and resource-efficient software and have been doing so over the past 15 years by delivering multiple bioinformatics tools, including GCTA, SMR, and OSCA.
First, GCTA (genome-wide complex trait analysis) was initially developed to estimate the proportion of variance in a phenotype that can be explained by all genome-wide SNPs (Yang et al. 2011 AJHG). It has been extended substantially to include modules for genome-wide association analysis (e.g., fastGWA, fastGWA-GLMM, MLMA, MLMA-LOCO), fine-mapping (COJO), gene-based tests (fastBAT, ACAT-V, and fastGWA-BB), and Mendelian randomization analysis (e.g., GSMR), etc.
Second, OSCA is a sister tool of GCTA, developed for estimating the proportion of phenotypic variance captured by all DNA methylation (DNAm) probes, LMM-based methylome-wide association analysis (i.e., testing the association of a probe conditioning on all the probes fitted as random effects), or methylation QTL mapping (Zhang et al. 2019 Genome Biol). Note that although developed primarily for DNAm data, OSCA is, in principle, applicable to all types of omics data, including gene expression and brain imaging data. Finally, SMR is a tool to implement the SMR and HEIDI methods to identify genes and/or functional genomic elements responsible for GWAS signals (Zhu et al. 2016 Nat Genet). To enhance the accessibility of this approach, we also launched the SMR-Portal, a user-friendly online platform for streamlined SMR analysis and visualization (Guo et al. 2025 Nat Metho).